芳烃和烯烃均含C(sp²)–H键,但二者的反应性截然不同------芳烃因自生芳香性作用发生亲电取代(如:Friedel–Crafts烷基化),而烯烃无法发生类似取代反应(图1a),与亲电试剂的反应通常会进行加成而非取代,因此尚无通用的方法来实现烯烃的直接烷基化。目前,化学家合成取代烯烃主要有三种策略:1)羰基化合物的烯烃化,如Wittig 反应、Horner–Wadsworth–Emmons(HWE)反应和 Julia 反应;2)炔烃的还原烷基化;3)烯烃交叉复分解反应,但受限于烯烃配对兼容性,并且无法用于1-烯烃/1,1-二取代烯烃的E-选择性交叉复分解,更不适用于三取代、环状等位阻烯烃。除此之外,烯基亲核试剂(如:烯基格氏试剂)或亲电试剂(如:烯基(拟)卤化物)与烷基片段之间的交叉偶联是合成烯烃的另一种策略,但前者制备繁琐、官能团耐受差;后者需多步合成(如:二溴化/消除),易产生多种结构异构体,实用性低。尽管先前已报道过烯基噻蒽鎓盐的烷基化反应,但仅适用于末端烯烃和简单的烷基锌试剂,而且其他烯基锍盐的烷基化仅限于苯乙烯。迄今为止,烯烃烷基化的方法普遍局限于活化烯烃(如:苯乙烯、迈克尔受体)且烷基的底物范围窄,同时Heck-烷基化也受限于苯乙烯或其它活化烯烃。另一方面,烷基羧酸储量丰富、结构多样、稳定易得,并且其与烯烃同为有机合成中最基础的官能团,但是将二者作为砌块直接构建C(sp²)–C(sp³)键的通用方法却尚未报道过。
近日,德国马克斯•普朗克煤炭研究所的Tobias Ritter教授课题组利用极性脱羧烷基化策略,成功实现了羧酸为烷基源的烯烃C–H键烷基化反应,并以优异的区域和非对映选择性获得多种烯烃(图 1b)。具体而言,氧化还原活性酯与锌原位生成持久性烷基锌中间体,经钯催化与烯基噻蒽鎓盐高效偶联便可获得一系列取代烯烃。该方法不仅操作简单,还适用于环状/链状、末端/内烯、单/双/三取代烯烃,并兼容多种结构复杂的烷基片段(图 1c)。此外,该策略规避了自由基偶联的化学选择性难题,拓展了难以合成的高取代烯烃的构建途径。相关成果发表在Nature 上。
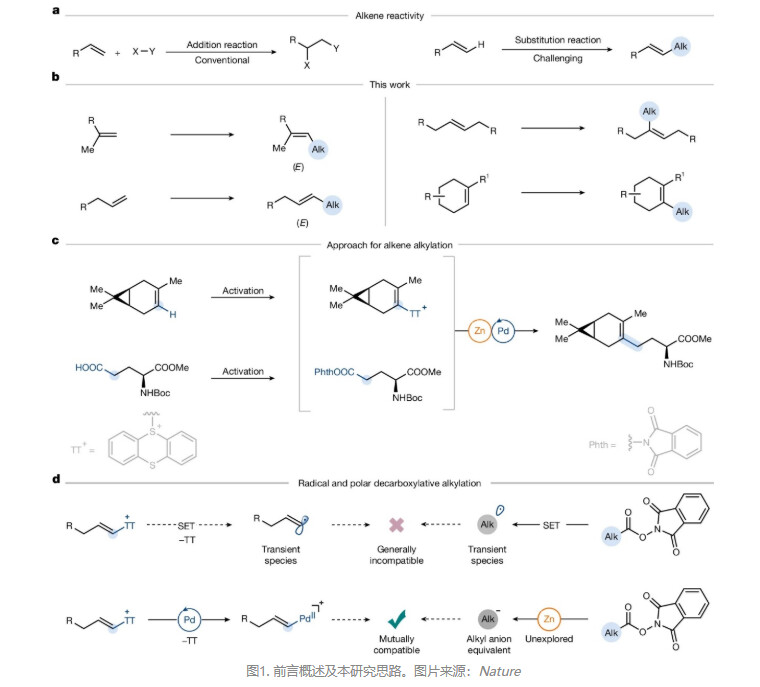
先前研究表明烯烃的C−H键可经加成–消除序列一步完成乙烯基C–H官能团化,从而直接将简单烯烃转化为高活性烯基亲电试剂(即烯基噻蒽鎓盐)。另外一方面,羧酸在转化为氧化还原活性酯后,其可作为通用的烷基源。然而,现有脱羧烷基化均经单电子路径生成烷基自由基,而烯基噻蒽鎓盐虽可被单电子还原(Ep = −1.68 V vs Ag/AgNO₃ in DMF),但两个瞬态自由基(烷基自由基与烯基自由基)的选择性交叉偶联面临根本性挑战(图1d)。进一步通过实验发现,在镍/铁催化单电子还原条件下主要观察到烯基噻蒽鎓盐原位脱噻蒽基化及氧化还原活性酯的加氢脱羧,进而表明双亲电体系下自由基路径存在严重化学选择性问题。基于此,作者转而探索极性双电子路径:即以Pd(0)/Pd(II)循环活化烯基噻蒽鎓盐,同时将氧化还原活性酯转化为碳负离子等价物。即烷基氧化还原活性酯与锌生成持久稳定的烷基锌中间体,其与烯基噻蒽鎓盐在钯催化下高效交叉偶联,从而高选择性构建C(sp²)–C(sp³)键(图1d)。
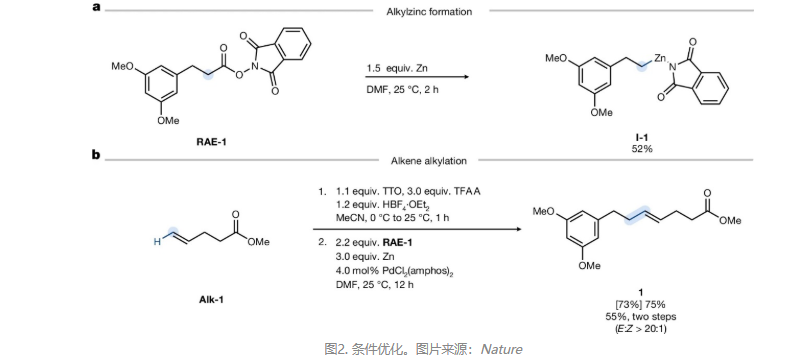
先前研究表明锌在乙腈或乙酸乙酯中还原氧化还原活性酯时可生成烷基自由基。然而,氧化还原活性酯RAE-1和Zn在DMF中反应2 h后,以52%的产率获得相应的烷基锌物种I-1(图2a)。相比之下,在THF或乙腈等常用偶联溶剂中,类似的锌还原无法生成具有合成适用性的烷基锌亲核试剂,作者推测首次单电子转移产生的烷基自由基在DMF中迅速与Zn⁺发生笼内重组,并被极性非质子环境稳定,从而导向烷基锌物种而非自由基产物。进一步向反应混合物中加入Pd催化剂和烯基噻蒽鎓盐,以75%的产率获得偶联产物1(图2b)。若所有组分一并加入,则主要发生脱噻蒽基质子化,这表明烷基锌形成速率慢于烯基噻蒽鎓盐对Pd(0)的氧化加成,故预生成烷基锌物种是高效偶联的前提。另外,对照实验表明羧酸原位制备的氧化还原活性酯也可以反应,但产率降低(40%);同时Mn、Mg及有机还原剂TDAE均无法替代Zn实现交叉偶联。
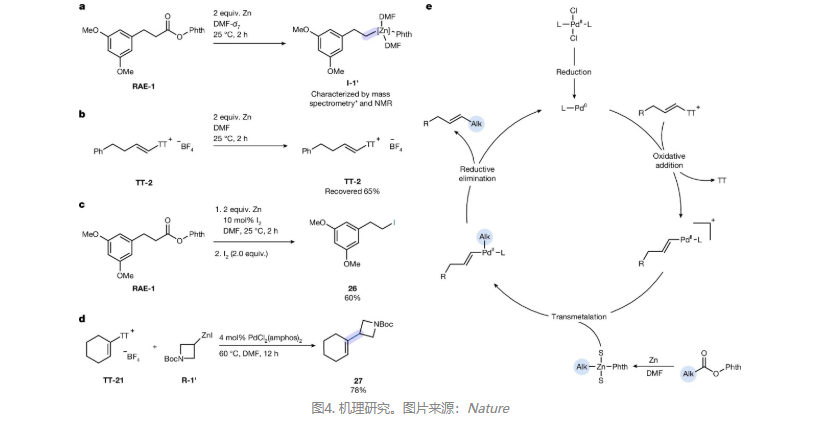
作者通过一系列实验对反应机理进行研究(图4),具体而言:1)氧化还原活性酯(Ep = −1.59 V vs Ag/AgNO₃ in DMF)与烯基噻蒽鎓盐(Ep =−1.68 V)的还原电位相近,但动力学分析显示锌对前者的还原速率显著快于后者(图4a、4b);2)反应中生成的可溶性Zn(II)对氧化还原活性酯与金属锌的反应具有自催化作用,外源烷基锌的加入可进一步加速化学选择性还原,这可能是通过Lewis酸配位活化氧化还原活性酯;3)在DMF-d₇中进行锌介导的RAE-1还原反应时,¹H/¹³C NMR谱证实形成了持久性烷基锌物种,同时ESI-MS分析检测到其与两分子DMF配位的加合物,这表明溶剂配位是稳定该中间体的关键。NMR分析还显示三种主要副产物:脱羧质子化生成的烷烃、未反应羧酸及同偶联二聚体,进而验证脱羧步骤经自由基路径发生。相比之下,在THF-d₈中进行反应时未观测到烷基锌信号;4)定量验证原位锌活化过程:向RAE-1和锌反应2 h后的混合物中加入I₂,以52%的产率得到RAE-1衍生的烷基碘。向标准烷基化反应中添加10 mol% I₂,产率提升8%(图4c);5)当使用预制碘代烷衍生的烷基锌试剂替代氧化还原活性酯进行反应时,在相同条件下以78%的产率获得目标偶联产物(图4d),进而证实原位形成的烷基锌为真实活性烷基化物种。在此基础上,作者提出了合理的反应机理(图4e):首先,PdII被还原为相应的Pd(0)物种,再与烯基噻蒽鎓盐进行氧化加成并得到相应的烯基-PdII物种,其与烷基锌试剂(由氧化还原活性酯与锌进行反应得到)进行转金属化并得到烯基,烷基-PdII物种,最后经还原消除便可得到所需的交叉偶联产物,同时再生Pd(0)物种。
Tobias Ritter教授课题组利用极性脱羧烷基化策略,成功实现了羧酸为烷基源的烯烃C–H烷基化,以优异的区域和非对映选择性获得多种烯烃。该方法不仅操作简单,还适用于环状/链状、末端/内烯、单/双/三取代烯烃,并兼容多种结构复杂的烷基片段。此外,该策略规避了自由基偶联的化学选择性难题,拓展了难以合成的高取代烯烃的构建途径。