药品有效期是基于原料药与制剂的稳定性研究数据,结合科学的试验设计、数据统计分析及风险评估形成的系统性结论。结合《普通口服固体制剂溶出度试验技术指导原则(2015 年)》《化学药物(原料药和制剂)稳定性研究技术指导原则(2015 年)》等技术文件,详细阐述药品有效期的制定逻辑、试验基础与核心流程。
一、药品有效期制定的核心基础:稳定性研究
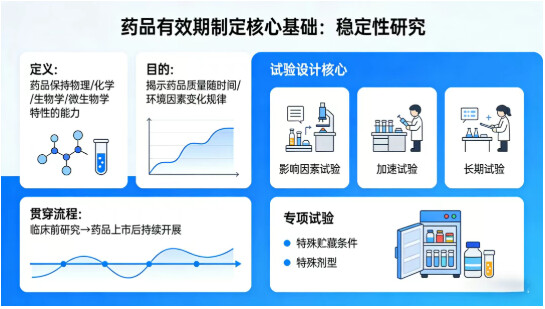
药品的稳定性是其保持物理、化学、生物学和微生物学特性的能力,而稳定性研究是揭示药品质量随时间、环境因素变化规律的关键,也是有效期制定的唯一科学依据。
稳定性研究贯穿药品研发全流程,从临床前研究开始,至药品上市后仍需持续开展,其试验设计围绕影响因素试验、加速试验、长期试验三大核心展开,同时针对特殊贮藏条件、特殊剂型增加专项试验。
(一)影响因素试验:明确药品稳定性特征
影响因素试验是在剧烈条件下开展的试验,原料药主要考察高温、高湿、光照、酸、碱、氧化等条件,制剂重点进行光稳定性试验,必要时增加低温、冻融试验。该试验仅需 1 个批次样品(结果不明确时加试 2 批),目的是明确药品对环境因素的敏感性、主要降解途径及降解产物,为加速试验、长期试验的条件设置,以及包装材料、贮藏条件的选择提供参考,同时验证质量检测方法的专属性。
(二)加速试验:模拟短期超常条件的质量变化
加速试验通过设置超出药品拟定贮藏条件的高温、高湿环境,考察药品在运输、贮藏环节中,短期偏离规定贮藏条件时的质量变化情况。该试验结果可作为长期试验条件设置的依据,同时能初步预测药品的长期稳定性。
试验统一采用 3 个批次样品开展,不同贮藏类型药品的试验条件与考察周期明确如下:
-
常规贮藏药品:试验条件为 40℃±2℃/75% RH±5% RH,考察周期 6 个月;
-
拟冷藏药品:试验条件为 25℃±2℃/60% RH±5% RH,考察周期 6 个月。
若加速试验过程中,药品质量出现显著变化,需额外增设中间条件试验,试验条件为 30℃±2℃/65% RH±5% RH,考察周期延长至 12 个月。
其中,制剂质量"显著变化" 明确为:含量与初始值相差 5% 或效价不符合规定;降解产物超出有效期标准;外观、物理性质、功能性试验不符合规定;pH 值不符合规定;12 个剂量单位的溶出度不符合规定;半渗透性容器包装制剂失水率达 5% 以上(小容量≤1mL 制剂除外)。原料药则以超出质量标准规定为 “显著变化”。
(三)长期试验:确定有效期的核心依据
长期试验是在药品拟定贮藏条件下开展的稳定性试验,直接反映药品在运输、保存、使用全过程中的质量变化,是制定有效期的最终依据。试验采用 3 个批次样品,根据药品贮藏要求设置不同条件:
-
常规贮藏药品:25℃±2℃/60% RH±5% RH 或 30℃±2℃/65% RH±5% RH,考察时间至少覆盖拟申请的有效期;
-
冷藏药品:5℃±3℃,考察至少 12 个月;
-
冷冻药品:-20℃±5℃,考察至少 12 个月;
-
半渗透性容器包装的水溶液制剂:25℃±2℃/40% RH±5% RH 或 30℃±2℃/35% RH±5% RH,重点考察失水性。
长期试验的取样频率需能准确反映药品质量变化趋势,有效期≥12 个月的药品,第一年每3个月取样一次,第二年每 6 个月一次,以后每年一次。注册申报时,新制剂需提供至少 3 个注册批次 12 个月的长期试验数据,仿制制剂需提供至少 3 个注册批次 6 个月的长期试验数据,同时需承诺继续考察至拟定有效期。
二、原料药与制剂有效期制定的特殊考量
(一)原料药的有效期(复检期)
多数稳定的化学原料药制定复检期,而非有效期。复检期是指原料药在规定贮藏条件下,可认为其符合质量标准并用于制剂生产的期限;复检期后若需使用,需重新复检,复检合格可立即使用,且可多次复检。原料药复检期的制定流程与制剂有效期一致,基于长期试验数据,结合降解趋势和批次间变异分析确定,标签上需注明复检日期。
(二)普通口服固体制剂的溶出度关联要求
普通口服固体制剂的溶出度是有效期制定的关键指标,需符合《普通口服固体制剂溶出度试验技术指导原则(2015 年)》要求:药品在有效期内的溶出度需与注册批次、关键临床试验批次的溶出曲线一致,若溶出特性在储存、运输中发生改变,需结合生物等效性试验结果重新评估有效期。
(三)特殊剂型与贮藏条件药品的有效期
-
半渗透性容器包装制剂:除考察常规指标外,重点控制失水性,在 40℃/≤25% RH 条件下放置 3 个月,失水量与初始值相差 5% 即为显著变化,需结合失水率数据调整有效期;
-
冷藏 / 冷冻药品:冷藏药品若加速试验前3个月质量显著变化,需评估短期偏离贮藏条件的影响,必要时缩短有效期;冷冻药品无规定加速试验条件,需通过略高温度下的试验了解质量变化,有效期完全基于长期试验实际数据确定,不得外推;
-
临用现配 / 多剂量包装制剂:需额外进行配伍稳定性或开启后使用稳定性试验,为配制后、开启后的使用期限提供依据,该期限也属于药品有效期体系的一部分。
三、药品有效期的标注要求
药品有效期的标注需遵循国家相关管理规定,基于对药品稳定性信息的全面评估,做到清晰、准确、规范:
-
贮藏条件的表述需具体,避免使用"室温"“环境条件” 等不确切词汇,如 “25℃以下密封保存”“5℃±3℃冷藏” 等,对不能冷冻的药品需特殊注明;
-
标签上需注明失效日期(制剂)或复检日期(原料药),失效日期为生产日期与有效期的加和,复检日期为生产日期与复检期的加和;
-
普通口服固体制剂等剂型,需在说明书中明确有效期内的关键质量指标要求(如溶出度),指导临床合理使用。